La fibrosis pulmonar idiopática (FPI), la forma más común de las neumonías intersticiales idiopáticas, es una enfermedad pulmonar crónica, progresiva, irreversible y letal, de causa desconocida. Se produce en personas de mediana edad y adultos de edad avanzada (edad media de diagnóstico 66 años, se limita a los pulmones y se asocia con un patrón histopatológico o radiológico típico de neumonía intersticial usual (NIU).
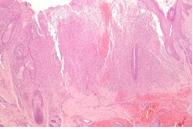
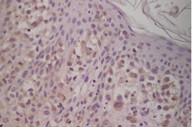
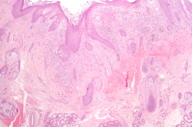
La característica histopatológica principal de la NIU, mejor vista con poco aumento, es el aspecto heterogéneo. Se observan áreas de fibrosis subpleural y paraseptal y panal de abejas (espacios aéreos de fibrosis quística revestidos por epitelio bronquiolar a menudo llenos de mucina y, un número variable de células inflamatorias) que alternan con zonas de parénquima menos afectado o normal (heterogeneidad espacial). En el fondo de los depósitos de colágeno se hallan pequeñas áreas de fibrosis activa (focos de fibroblastos) que reflejan la heterogeneidad temporal del proceso patológico e indican las actividades actuales.
En general, la inflamación es leve y consiste en un infiltrado intersticial linfoplasmocitario desigual. Los signos de la tomografía computarizada (TC) de alta resolución representan el patrón de la NIU, caracterizado por opacidades reticulares a menudo asociadas con bronquiectasias de tracción, con poca o ninguna opacificación en vidrio esmerilado. Es común la presencia de lesiones subpleurales en panal de abejas, con espacios aéreos quísticos agrupados con paredes bien definidas (por lo general de 3-10 mm de diámetro). La presencia de estas lesiones es fundamental para hacer un diagnóstico definitivo.
Los pacientes con FPI generalmente solicitan atención médica porque sufren disnea de esfuerzo y tos crónicas y progresivas. La auscultación pulmonar halla con frecuencia estertores crepitantes inspiratorios bibasales; se constatan dedos en palillo de tambor. La historia natural de la FPI se caracteriza por ser un trastorno pulmonar estable o lentamente progresivo, patrón que siguen la mayoría de los pacientes. Sin embargo, los últimos hallazgos indican que la FPI es un grupo heterogéneo enfermedades y se están describiendo nuevos fenotipos clínicos con distintos patrones de supervivencia.
Los mecanismos patogénicos no están claros, pero cada vez hay más evidencia que indica que la enfermedad es el resultado de un comportamiento anormal de las células del epitelio alveolar que provoca la migración, la proliferación y la activación de las células del mesénquima, con la formación de focos de fibroblastos y miofibroblastos. Los miofibroblastos activados secretan cantidades exageradas de moléculas de la matriz extracelular con la destrucción posterior de la arquitectura pulmonar.
Epidemiología y factores de riesgo
La incidencia anual de la FPI está aumentando y se estima de 4,6 a 16,3 por 100.000 personas, con una prevalencia de 13 a 20 casos cada 100 000. La enfermedad predomina más en los hombres (1,5-1,7:1) y su frecuencia aumenta con la edad. Los factores de riesgo ambientales importantes son el tabaquismo y la exposición al polvo de metales y madera. La transmisión genética se produce en alrededor de 0,5-3,7% de los pacientes con FPI, aunque esta frecuencia puede ser mayor en las familias afectadas con un patrón de transmisión vertical autosómico dominante con reducción de la penetrancia. En general, los casos familiares de FPI se pierden. Todavía falta definir el efecto sobre la evolución clínica de la FPI de varias condiciones comórbidas - obesidad, diabetes mellitas, reflujo gastroesofágico, hipertensión pulmonar, apnea obstructiva del sueño, enfermedad arterial coronaria, y enfisema.
Diagnóstico
El diagnóstico de FPI a menudo requiere el enfoque multidisciplinario de neumólogos, radiólogos y anatomopatólogos especializados en el campo de las enfermedades pulmonares intersticiales. Un patrón indicativo de NIU en la TC de alta resolución o el tejido pulmonar obtenido por biopsia quirúrgica del pulmón es crucial para el diagnóstico final. Los diagnósticos diferenciales principales son, en primer lugar, la neumonía intersticial inespecífica fibrótica seguido por el de otras formas de neumonías intersticiales idiopáticas y enfermedades intersticiales pulmonares ocupacionales o ambientales, enfermedades sistémicas y fármacos.
Se recomienda la evaluación serológica de las enfermedades del tejido conjuntivo, incluso en ausencia de signos o síntomas de ellas. Todavía no se cuenta con biomarcadores fiables de muestras de sangre o del líquido del lavado broncoalveolar que podrían ser útiles para el diagnóstico diferencial o la predicción de los resultados.
Fenotipos clínicos y pronóstico
La FPI tiene un curso clínico heterogéneo, y los pacientes tienen una supervivencia media de 2,5-3,5 años a partir del diagnóstico. Se han definido fenotipos clínicos con distintos patrones de comorbilidades y supervivencia. El peor pronóstico se asocia con la edad (>70 años), el antecedente de tabaquismo, un índice de masa corporal bajo, el deterioro fisiológico grave, la gran extensión radiológica de la enfermedad y la hipertensión pulmonar.
Evolución estable o lentamente progresiva
Muchos pacientes con FPI tienen una evolución clínica relativamente lenta y por lo general consultan al médico meses o años después del comienzo de los síntomas (tos y disnea progresivas). En la presentación, los pacientes tienen disminución del volumen y la capacidad pulmonar, con hipoxemia en reposo que empeora con el ejercicio. En los grupos de placebo de los grandes ensayos clínicos, la tasa media anual de disminución de la capacidad vital forzada oscila entre 0,13 y 0,21L.
Evolución acelerada
Un subgrupo de pacientes, los fumadores de cigarrillos, en su mayoría hombres, tienen un curso rápidamente progresivo con una supervivencia más corta; esta variante es conocida como FPI acelerada. En estos casos, la señal de transcripción indica la regulación hacia arriba de varias vías funcionales, que en su mayoría operan en los dominios epiteliales y mesenquimales alveolares. La FPI acelerada se diferencia de la FPI típica de progresión lenta en el curso clínico y el perfil de transcripción, a pesar de que en el momento del diagnóstico tienen una función pulmonar y signos en la radiografía de tórax similares.
Exacerbación aguda
La exacerbación aguda de la FPI está definida por el deterioro rápido en ausencia de infección, insuficiencia cardiaca, embolia pulmonar u otra causa identificada. El diagnóstico se realiza mediante una combinación de signos clínicos (empeoramiento e la disnea a los pocos días o semanas), hallazgos fisiológicos (reducción severa de la PaO2 en la sangre arterial) y radiográficos (imágenes en vidrio esmerilado bilaterales y consolidación en superposición con un patrón típico de NIU en la TC de alta resolución). Se estima que las exacerbaciones agudas de la FPI se presentan en el 5-20% de los casos.
Los pacientes con esta exacerbación aguda evolucionan mal, con una mortalidad superior al 60% durante la internación hospitalaria, y entre los que sobreviven hay una mortalidad >90% a los 6 meses después del alta. Durante los periodos de exacerbación se ha detectado el virus Torque Teno en el 27% de los casos de FPI. Morfológicamente, se puede observar daño alveolar difuso superpuesto a las características típicas de la NIU. Los mecanismos patogénicos son desconocidos pero se ha informado la presencia de apoptosis epitelial durante los periodos de exacerbación aguda. Durante las exacerbaciones agudas podrían estar en juego esas células porque su número aumenta y vuelve a los niveles anteriores a la exacerbación cuando el paciente se recupera.
Fibrosis pulmonar y otros trastornos pulmonares
El diagnóstico de fibrosis pulmonar combinada con enfisema se basa en la TC de alta resolución que muestra lesiones enfisematosas en los lóbulos superiores y lesiones similares a las de la NIU en los lóbulos inferiores. Si la fibrosis pulmonar combinada con enfisema es una condición clínica distinta, un fenotipo clínico de FIP diferente en los fumadores, especialmente los grandes fumadores, o corresponde a dos enfermedades paralelas distintas, todavía no ha quedado establecido. Estos pacientes desarrollan hipertensión arterial pulmonar temprana y grave y tienen menor supervivencia comparados con los pacientes con FPI sin enfisema.
La fibrosis pulmonar combinada y la hipertensión pulmonar tienen un efecto pronóstico negativo en los pacientes con FPI y se asocia con baja capacidad de difusión del monóxido de carbono, menor distancia en la caminata de 6 minutos, desaturación durante el ejercicio y aumento del riesgo de muerte.
El carcinoma broncogénico es común en los pacientes con FPI (9,8-38%) pero el mecanismo se desconoce. Sin embargo, hay una asociación con el tabaquismo; la mayoría de los cánceres de pulmón en los pacientes con fibrosis pulmonar combinada y cáncer están en las zonas periféricas con fibrosis y anomalías epiteliales graves, lo que implica que el proceso fibrótico es el mismo que en la patogénesis del cáncer de pulmón.
Patogénesis
Desde la enfermedad inflamatoria hasta la enfermedad por alteración epitelial
La inflamación tiene un papel fundamental en la mayoría de las enfermedades intersticiales del pulmón y, si es crónica, evoluciona a la fibrosis. Sin embargo, con la redefinición de FPI como una enfermedad distinta caracterizada por un patrón típico de NIU, la reacción fibrótica progresiva de la FPI se asocia con un proceso activado por los fibroblastos dependientes del epitelio y una escasa respuesta al tratamiento con antiinflamatorios. Sin embargo, en un subgrupo de pacientes, la desregulación de los mecanismos inmunes adaptativos y la inflamación posterior podrían representar un papel en el comienzo o la progresión de la enfermedad. Por lo tanto, la fibrosis pulmonar epitelial podría tener al menos dos rutas celulares diferentes - la vía inflamatoria y la vía epitelial, las que podrían dar lugar a la fibrosis pulmonar.
Lesiones epiteliales y activación: interacciones genéticas y ambientales
Varios factores ambientales pueden contribuir a las lesiones epiteliales y la apoptosis, como el tabaquismo y la microaspiración crónica silenciosa. Por otra parte, la infección viral crónica, principalmente por el virus herpes, podría contribuir a la patogénesis de la FPI.
No hay factores genéticos asociados consistentemente con la FPI esporádica. En algunos casos familiares de fibrosis pulmonar que tienen mutaciones en la proteína surfactante C aparecen alteraciones en la respuesta de la proteína no plegada, una proteína hidrofóbica expresada exclusivamente por células alveolares epiteliales activadas tipo II (CEA II), activadas en forma aberrante. El cambio de sentido o las mutaciones en la deleción corta de esta proteína provocan la producción de proteínas mal plegadas, que, al acumularse o agruparse en complejos pueden causar la lesión de las células epiteliales. Un polimorfismo común en la región promotora del gen de la mucina 5B (MUC5B) se asocia con la neumonía intersticial familiar y la FP esporádica.
El estudio del genoma de 6 familias con FPI familiar mostró un haplotipo compartido en el cromosoma 4q31, que fue significativamente más frecuente en los pacientes que en los controles de la población. Este haplotipo albergaba ELMOD2, un gen expresado en el pulmón, pero cuya expresión es significativamente menor en el pulmón de la FPI, comparado con la de los pulmones sanos. El ELMOD2 es esencial para los procesos celulares y podría tener un efecto antiviral en las CEA. También se han identificado mutaciones de la telomerasa.
A pesar de la lesión epitelial y la apoptosis, hay un aumento del número de neumocitos tipo II hiperplásicos e hipertróficos, una característica notable de los pulmones afectados por la FPI. Por otra parte, se observan células epiteliales grandes y alargadas o atenuados. También se ha informado la presencia de epitelio tipo bronquiolar y de metaplasia escamosa revistiendo las lesiones en panal de abejas. Las células epiteliales son muy activas, lo que lleva a la desregulación del proceso de reparación que parece estar perpetuamente activado, incluso en ausencia del estímulo primario.
Nuevas pruebas indican que la desregulación de algunas vías embriológicas podría explicar el comportamiento anormal del CEA y tal vez de los fibroblastos. Los Ligandos Wnt forman una gran familia que secreta glucoproteínas esenciales para los procesos morfogenéticos. Los resultados de varios estudios indican que el epitelio alveolar y los fibroblastos expresan en exceso a miembros de la vía Wnt/Wg en los pulmones de la FPI. Por otra parte, hay una gran acumulación nuclear de catenina β en las CEA y los fibroblastos, lo que sugiere que en ambos tipos celulares la vía Wnt-β-catenina está activada.
El gen homólogo de la fosfatasa y la tensina (PTEN) es crucial para el desarrollo. En los adultos, participa en la regulación de los procesos fisiológicos tales como la polaridad, la proliferación, y la apoptosis de las células. En los pacientes con FPI, la expresión de PTEN está regulada hacia abajo en los miofibroblastos dentro de los focos fibroblástcios, lo que podría explicar su supuesta resistencia a la apoptosis. La interacción de la integrina ß1-colágeno en los fibroblastos normales activa al PTEN, que es también un regulador negativo del crecimiento, mientras que el mecanismo de retroalimentación negativo es defectuoso en los fibroblastos de la FPI.
El gen sonic hedgehog (Shh) es un morfogen esencial para la definición de los patrones durante la embriogénesis. Este ligando del desarrollo permite que las células evadan la apoptosis y la detención del ciclo celular, favoreciendo la proliferación. En los pulmones afectados por la FPI hay una fuerte expresión de Shh, principalmente en las células epiteliales que recubren los quistes de la lesión en panal de abejas.
Las proteínas morfogenéticas óseas pertenecen a la superfamilia del factor de transformación del crecimiento β (TGF β) y tienen un papel esencial en el estado embrionario y el desarrollo posnatal. En los adultos, la reactivación de la expresión de los antagonistas de la proteína morfogenética ósea puede contribuir a la progresión de algunas enfermedades crónicas degenerativas.
Se ha informado que en los fibroblastos de los pulmones de la FPI existe un aumento de la expresión de gremlina, un fuente antagonista de una proteína morfogenética ósea. El aumento de las concentraciones de gremlina podría atenuar la fosforilación mediada por la señalización de la proteína morfogenética ósea en los pulmones, dando lugar a la transición epitelio-mesénquima inducida por el TGFβ1 y a la disminución de la apoptosis de los miofibroblastos.
Efectos profibróticos de las CEA en forma aberrante en el microambiente pulmonar
Un proceso patológico importante en la FPI es la activación de la cascada de la coagulación, que tiene varios efectos profibróticos. En la FPI, el complejo de factores tisulares-factor VIIa-Factor X se forma en el epitelio alveolar, permitiendo la activación del factor X que a su vez estimula los fibroblastos en las regiones fibróticas subyacentes. La matriz provisional, formada por fibrina y fibronectina, podría estimular la transición epitelio-mesénquima, incluso en ausencia de TGFβ1. Por otra parte, la trombina y el factor X activados inducen la diferenciación de los fibroblastos pulmonares a miofibroblastos a través de la vía del receptor 1 activado por la proteasa. Estos hallazgos proporcionan pruebas convincentes de que en la FPI se activa la señalización procoagulante y que la función de la fibrinólisis alveolar deficiente, causada principalmente por las células epiteliales, representa un papel importante en la respuesta fibrótica del pulmón.
En los tejidos dañados, los fibroblastos se activan y diferencian en miofibroblastos, los cuales son células contráctiles especializadas con mayor potencial profibrótico que los fibroblastos. En los focos fibroblásticos, estas células provocan el depósito exagerado de matriz extracelular, que es el sello distintivo del proceso de cicatrización que conduce a la destrucción de la arquitectura pulmonar. Se desconoce el origen de los fibroblastos y los miofibroblastos y las razones por las cuales en la FPI se organizan en focos morfológicamente distintos.
Hay muchas pruebas de que las CEA son la fuente principal de los mediadores que funcionan como factores quimiotácticos o mitógenos de las células del mesénquima, como el factor de crecimiento derivado de las plaquetas, el factor de necrosis tumoral α (TGF α) y la endotelina 1. Estos factores probablemente contribuyen sobre todo a la migración, la proliferación y la diferenciación de las células del mesénquima.
Hay un influjo de circulación de fibrocitos en los pulmones de la FPI, los que constituyen una subpoblación particular de leucocitos caracterizados porque expresan marcadores de células hematopoyéticas (CD45, CD34) y mesenquimales (colágeno I, fibronectina). La mayoría de los fibrocitos circulantes expresan el receptor de quimiocinas CXCR4, lo que sugiere que el eje CXCR4-CXCL12 es crucial para el traslado en los pulmones de la FPI. Las CEA de estos pacientes expresan gran cantidad de CXCL12, lo que probablemente constituye el gradiente quimiotáctico necesario para el tráfico de fibrocitos positivos para CXCR4.
El epitelio podría contribuir directamente a la expansión de la población de fibroblastos y miofibroblastos a través de la transición epitelio-mesénquima. En este proceso, las células epiteliales adquieren propiedades mesenquimáticas por las cuales aumentan su capacidad para moverse y sintetizar matriz intersticial. En la FPI, las células mesenquimáticas locales, los fibrocitos circulantes y la transición epitelio-mesénquima participan en la expansión de los fibroblastos y los miofibroblastos. Sin embargo, no se sabe en qué medida participa cada uno en el inicio o la perpetuación de la enfermedad.
Diferenciación de los fibroblastos en miofibroblastos
En esta diferenciación intervienen 3 factores principales: la tensión mecánica (que induce la diferenciación de los protomiofibroblastos), el aumento local de la actividad del TGFβ1 y la presencia de proteínas especializadas de la matriz. Los miofibroblastos causan una acumulación adicional exagerada de matriz celular (fibrosis) y contribuyen a la interrupción de la membrana basal y la muerte de las células epiteliales.
La eliminación de los miofibroblastos por la apoptosis es esencial durante el proceso normal de cicatrización, que no parece producirse en los focos fibroblásticos de la FPI. Las señales del microambiente como el TGFβ1 y la endotelina 1 (en su mayoría sintetizados por las CEA en la FPI) podrían promover la resistencia a la apoptosis de los fibroblastos a través de las vías de señalización que involucran PI3K/AKT. Sin embargo, no hay evidencia convincente de que la reducción de la susceptibilidad a la apoptosis ocasiones la persistencia de los miofibroblastos in vivo. Por otra parte, se desconocen las razones por las cuales aparentemente los fibroblastos y los miofibroblastos sobreviven mientras que las células epiteliales mueren dentro del mismo microentorno. Una explicación posible sería la deficiencia de prostaglandina E2 que se observa en la FPI, que aumenta la sensibilidad de las CEA a la apoptosis pero la disminuye en los fibroblastos.
Ausencia de neumocitos tipo I
Las CEA tipo I cubren más del 90% de la superficie alveolar en la periferia del pulmón, y la interconexión con los capilares pulmonares proporciona una superficie fácilmente permeable para los gases. Los pacientes con FPI tienen una importante pérdida de neumocitos tipo I, aunque el efecto putativo de este proceso patológico en la respuesta fibrótica no está claro. Por otra parte, la transdiferenciación de los neumocitos tipo II en neumocitos tipo I, imprescindible para volver a establecer un epitelio alveolar funcional, está profundamente alterada en la FPI, debido a las anormalidades severas de la matriz extracelular y la membrana basal del epitelio interrumpida.
La pérdida de las CEA tipo I podría provocar la reducción de algunas moléculas antifibróticas importantes (por ej. la caveolina-1). Sin embargo, no se sabe si en la patogénesis de la FPI está involucrada la disminución de las concentraciones de caveolina-1, específicamente atribuible a la pérdida de los neumocitos tipo I. Por otra parte, el receptor de los productos finales de la glicación avanzada (RPFGA) también está disminuido en la FPI. El RPFGA es un miembro de la superfamilia de inmunoglobulinas de los receptores de la superficie celular que son altamente expresados por las CEA tipo I normales. La pérdida de este receptor podría resultar en una disminución de la unión de las CEA tipo I a la membrana basal, lo que impide la correcta reepitelización de los alvéolos durante la fibrogénesis.
Metaloproteinasas de la matriz y remodelación pulmonar anormal
Entre los genes mayormente expresados en la FPI están las metaloproteinasas 7 (MMP7), MMP1 y MMP2 pero también se han reportado otras MMP en el líquido del lavado broncoalveolar (MMP3, MMP8 y MMP9) o se han localizado mediante técnicas de inmunohistoquímica (por ej., MMP de membrana). La mayoría de estas MMP se localizan en las CEA, pero pocas están en los focos fibroblásticos. La MMP1 se localiza principalmente en el epitelio y está casi ausente en el compartimiento intersticial, donde se acumula el colágeno. La MMP7 y la osteopontina interactúan para afectar el fenotipo de la FPI (por ej., MMP7 es inducida por la osteopontina y la osteopontina también se escinde y es activada por la MMP7).
Tanto la MMP1 como la MMP7 facilitan la migración de las células epiteliales, reduciendo la afinidad de la integrina α2β1. El aumento de la expresión de MMP1 podría estar en parte asociado a dos polimorfismos del gen que afecta la capacidad de respuesta a la transcripción. Las concentraciones de MMP2 están aumentadas tanto en las CEA alveolares como bronquiolares y en los focos fibroblásticos. Los principales activadores de la proMMP2 son miembros de la familia MMP de la membrana que también se expresa fuertemente en los pulmones de la FPI. Por lo tanto, la mayor cantidad de MMP2 activa podría ser atribuida a la acción de estas enzimas. La MMP9 también está elevada en la FPI, y un aumento de su forma activa en el lavado broncoalveolar se asocia con un fenotipo clínico acelerado.
Angiogénesis y remodelación vascular
La neovascularización es un proceso fundamental en el tejido de reparación después de una lesión y depende del equilibrio entre diversos factores, principalmente las quimiocinas que promueven o inhiben la angiogénesis. No se conoce cuál es el papel de la angiogénesis el pulmón de la fibrosis pulmonar. En los pulmones se produce una remodelación vascular aberrante, pero las áreas fibróticas tienen menos vasos sanguíneos mientras que el tejido de las zonas no fibróticas está muy vascularizado. Casi no hay capilares dentro de los focos fibroblásticos, lo que indica que el proceso fibrótico en la FPI no necesita la neovascularización. Por lo tanto, en ciertas configuraciones patológicas, el aumento de la angiogénesis parece tener un papel fibrogénico mientras que en otras, la angiogénesis favorece la fibrosis.
Procesos que vinculan el envejecimiento con la FPI
Los mecanismos que vinculan el envejecimiento con la FPI se desconocen. En general, la TC de alta resolución muestra signos en los individuos asintomáticos mayores (≥75 años) pero no en los más jóvenes. Un posible mecanismo está relacionado con un acortamiento acelerado de los telómeros, los que en cada división celular se acortan al alcanzar una longitud crítica, luego activan un sitio dependiente de p53 que conduce a la apoptosis o senescencia replicativa. Se cree que los telómeros cortos comprometen la replicación de las células progenitoras que se quedan en los tejidos después de la lesión. Las personas podrían estar en mayor riesgo de desarrollar FPI. Algunos pacientes con otras formas de FPI también tienen telómeros acortados, aumentando la posibilidad de mecanismos compartidos.
Sin embargo, el acortamiento de los telómeros también se observa en la enfermedad pulmonar obstructiva crónica, otra enfermedad relacionada con la edad pero completamente diferente. Por otra parte, se ha observado que los ratones con deficiencia de telomerasa que tienen un acortamiento secuencial espontáneo de los telómeros desarrollan lesiones enfisematosas.
Las CEA de esos ratones activan la vía de la respuesta al estrés y la apaptosis espontánea, indicando que el acortamiento de los telómeros provoca daño de las células epiteliales pero la consecuencia es el enfisema y no la fibrosis. Adicionalmente, en el enfisema humano también se ha observado apoptosis epitelial alveolar. No obstante, el cambio en la población total en la distribución de los telómeros sugiere que el acortamiento de los telómeros puede ser un cofactor patógeno para la FPI.
El envejecimiento también se asocia con el aumento del estrés oxidativo como resultado del desequilibrio entre los prooxidantes (especies de O2 reactivo y nitrógeno) y antioxidantes (por ej., superóxido dismutasa, glutatión). Las consecuencias son el daño directo del ADN, la oxidación de los ácidos grasos poliinsaturados en las membranas celulares y la inactivación de las enzimas. El estrés oxidativo excesivo tiene varios efectos nocivos que pueden contribuir a la patogénesis de la FPI, incluyendo la activación de vías de la vía de señalización redox-sensible, los cambios en la expresión de citocinas o quimiocinas, la modificación del balance las proteasas o antiproteasas, la inducción de la apoptosis y la activación de los fibroblastos.
Los procesos epigenéticos incluyen alteraciones transmisibles en la expresión genética causadas por otros mecanismos diferentes de los cambios en la secuencia del ADN, y estos procesos son esenciales para el normal desarrollo y mantenimiento de los patrones de expresión de los genes titulares específicos. Los mecanismos epigenéticos más comunes son la metilación del ADN, las modificaciones postraducción de las histonas y los efectos de transcripción de las moléculas no codificantes del ARN, como la microRNA.
En los ancianos (>65 años) hay una pérdida progresiva de la metilación del ADN en elementos repetitivos dispersos de todo el genoma que parece ser proporcional a la expectativa de vida. En el inicio y la progresión del cáncer interviene una reprogramación aberrante del epigenoma. Los cambios epigenéticos contribuyen al comportamiento agresivo de los fibroblastos en la FPI. Thy1, un receptor que inhibe la diferenciación de los fibroblastos en miofibroblastos y que disminuye la actividad fibrogénica, no se expresa en los focos fibroblásticos in vivo.
La pérdida de este receptor ocurre por el silenciamiento epigenético secundario a la hipermetilación de la islas de citosina-guanina en el promotor del gen. En comparación con los fibroblastos normales, los fibroblastos de la FPI expresan menos ciclooxigenasa 2 y sintetizan menos prostaglandina E2, un regulador hacia abajo potente de la activación de los fibroblastos. La menor expresión de ciclooxigenasa-2 en los fibroblastos de la FPI parece estar causada por anormalidades epigenéticas de la acetilación de las histonas que inhibe los factores de transcripción activados de la unión al promotor 2 de la ciclooxigenasa.
Los microARN son reguladores postranscripción que se unen a secuencias específicas, bloqueando la traducción o causando la degradación del ARN mensajero diana, lo que resulta en el silenciamiento de los genes. Las microARN probablemente controlan a la mayoría de las rutas y redes biológicas. Durante el envejecimiento, la desregulación de la expresión de las microRNA se produce generalmente en grupos, lo que indica que las acciones podrían estar funcionalmente coordinadas juntas por los reguladores de transcripción comunes.
La disrupción de estas alianzas vitales dependientes de la edad podría contribuir al desarrollo de las enfermedades en las personas de edad avanzada. Como ejemplo, la regulación negativa de algunas micro ARN (miR1, mirR30), junto con la regulación hacia arriba de otras (miR20a, miR21) son frecuentes en los pacientes con hipertrofia cardiaca y arteriosclerosis coronaria. Las microARN se expresan en la FPI en forma diferente que en los pulmones normales, con una disminución significativa de microARN let-7d. La inhibición de let-7d in vitro indujo la transición epitelio-mesénquima mientras que la inhibición in vivo causó fibrosis alveolar septal. Del mismo modo, en la FPI, los pulmones tienen una regulación hacia arriba de la miR21 y una regulación hacia abajo de la miR29, principalmente localizadas en los miofibroblastos.
El aumento de los niveles de miR21 promovió la actividad fibrogénica de TGFβ1 en los fibroblastos, mientras que la regulación hacia debajo de miR-21 atenuó esta actividad. Por el contrario, los niveles de miR29 se correlacionan inversamente con la expresión de varios genes profibróticos diana y con la gravedad de la fibrosis. Por lo tanto, la desregulación epigenética que influye en el fenotipo del envejecimiento podría aumentar el riesgo de desarrollar FPI si se alcanza un umbral crucial de epimutaciones.
Enfoques terapéuticos
En los ensayos clínicos de nuevos fármacos (etanercept, IFNx, bosentan, mesilato de imatinib) en pacientes con FPI con deterioro funcional leve a moderado no se han observado beneficios significativos. En general, los pacientes con FPI con deterioro funcional moderado a grave y comorbilidades asociadas (hipertensión pulmonar) han sido excluidos de los ensayos. En consecuencia, muchos pacientes atendidos en la práctica clínica no han sido estudiados. Están en desarrollo vario ensayos clínicos.
Con varias terapias nuevas, hay evidencia que sugiere beneficiosos clínicos en pacientes con FPI. La N-acetilcisteína, un antioxidante que se utiliza combinado con prednisona y azatioprina, reduce la tasa de disminución de la capacidad vital forzada y la capacidad de difusión del monóxido de carbono después de 12 meses de tratamiento. Sin embargo, los cambios observados son de significado clínico incierto. La pirfenidona, una compuesto nuevo que inhibe el TGF in vitro, disminuyó la tasa de declinación de la capacidad vital y aumentó el tiempo de supervivencia libre de progresión. Los datos combinados de 2 ensayos de FPI simultáneos en fase 3 (CAPACITY1 y 2) indican que la variación media de la capacidad vital forzada y la distancia en la caminata de 6 minutos disminuyeron en el grupo tratado con pirfenidona.
En un ensayo no ciego de FPI, los pacientes tratados con prednisolona y anticoagulantes (coumadin en pacientes ambulatorios y dalteparina intravenosa al ingresar al hospital) mejoraron la supervivencia global a los 3 años y la mortalidad asociada con la exacerbación aguda (63% vs. 35%) disminuyó en comparación con los pacientes tratados con prednisolona sola. Un estudio con warfarina para pacientes con FPI fue suspendido por la elevada posibilidad de que su efecto no fuera muy superior al del placebo
Trasplante de pulmón
El trasplante de pulmón es el único tratamiento que prolonga la supervivencia en la FPI avanzada, aunque la supervivencia postrasplante a los 5 años es de alrededor del 44%. El momento apropiado para remitir al paciente para el trasplante es muy controvertido dado el curso variable de la FPI y la falta de medidas de pronóstico validadas. Los pacientes con FPI suelen consultar tarde en el curso de su enfermedad y muchos mueren antes de recibir un trasplante. Por lo tanto, sería razonable derivar a los pacientes para trasplante apenas realizado el diagnóstico.
Tratamiento con células madre
Tanto las células embrionarias como las células madre derivadas del tejido adulto pueden participar en la regeneración y reparación de los órganos de los adultos enfermos, incluyendo los pulmones. El principal problema después de la lesión es restablecer la integridad y la organización funcional de la capa epitelial y las unidades alveolocapilares. Este proceso probablemente se produce durante la reparación normal por la migración y la difusión de las células progenitoras circulantes vecinas y recién reclutadas que proliferan y se diferencian fenotípicamente para cubrir las superficies desnudas.
Las células madre mesenquimales son una perspectiva prometedora para la regeneración tisular. En los ratones con lesión pulmonar por bleomicina, estas células emigran al pulmón, adoptan un fenotipo similar al epitelio y reducen la fibrosis. Por otra parte, después de la inyección intratraqueal de bleomicina, las células progenitoras epiteliales positivas para prominina 1 CD133 coexpresan marcadores de células madre y hematopoyéticas y se injertan en los pulmones, se diferencian en neumocitos tipo II y protegen contra la fibrosis inducida por bleomicina. Más recientemente, en el mismo modelo de ratón se estudió el potencial terapéutico de las CEA tipo II derivadas de células madre embrionarias humanas. Estas células diferenciadas se diferenciaron en neumocitos tipo I y anularon la respuesta inflamatoria y fibrótica. Por desgracia, casi todos los experimentos se han hecho en el modelo de pulmón lesionado por bleomicina, en el que la inflamación y la fibrosis son escasas y espontáneamente reversibles y no representan a la naturaleza progresiva y letal de la FPI.
Otra área de crecimiento de la investigación es el pulmón de bioingeniería usando tejido pulmonar en el que se cambiaron las células naturales por células del epitelio pulmonar neonatal y del endotelio microvascular del pulmón. Cuando se trasplantaron, estos pulmones fueron efectivos para el intercambio de oxígeno y del dióxido de carbono. Aunque se está lejos de la bioingeniería humana del pulmón, este enfoque es alentador.
Otros factores de manejo
Los pacientes con exacerbaciones agudas suelen tratarse con antibióticos de amplio espectro y corticoides. A menudo es necesaria la ventilación mecánica, pero por lo general no tiene buenos resultados, con una tasa elevada de mortalidad hospitalaria. Entre los pacientes que sobreviven y son dados de alta del hospital, la recurrencia es común y generalmente fatal. Los pacientes con FPI e hipertensión pulmonar tienen mayor mortalidad. En consecuencia, la terapia contra la hipertensión arterial pulmonar brinda beneficios. El sildenafil, un bloqueante de la fosfodiesterasa 5 en las áreas de pulmón bien ventiladas, reduce la resistencia vascular pulmonar y mejora el intercambio gaseoso en los pacientes con fibrosis pulmonar grave. El sildenafil puede mejorar mucho la disnea y la calidad de vida de los pacientes con FPI.
Aunque la asociación patogénica potencial entre la microaspiración crónica y la FPI sigue sin estar clara, hay algunas pruebas que apoyan el manejo del reflujo gastroesofágico. Sin embargo, se necesitan más estudios para comprobar si el tratamiento agresivo de esta enfermedad crónica es capaz de mejorar o detener su progresión. Teniendo en cuenta la relación entre el envejecimiento y la FPI, los médicos deben prestar mayor atención a las comorbilidades geriátricas y al manejo de los síntomas para complementar las nuevas terapias modificadoras de la enfermedad y mejorar la calidad de vida.
La rehabilitación pulmonar, los programas educativos y la formación de grupos de apoyo pueden ayudar a los pacientes a respirar mejor y realizar a sus actividades de la vida diaria con menos dificultad respiratoria. Para tratar la hipoxemia que tiende a empeorar con el ejercicio suele ser necesaria la oxigenoterapia.